IEC 62304 to FDA eSTAR: A Software Documentation Guide
Map IEC 62304 processes to FDA eSTAR requirements. Complete guide to 10 required software documents for medical device submissions with examples.
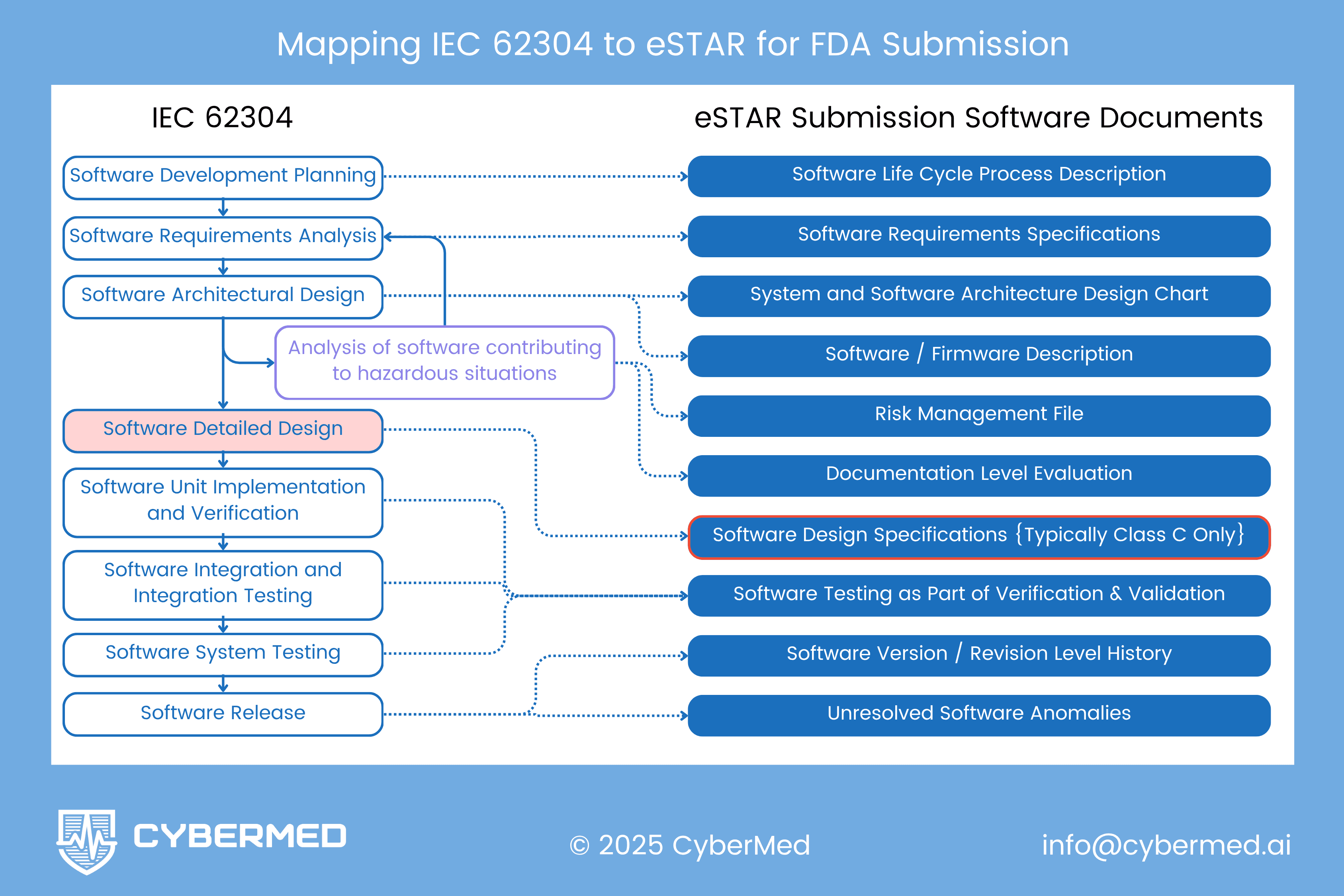
IEC 62304's development process maps directly onto the software documents FDA's eSTAR form requires: 9 documents for Basic documentation level, 10 for Enhanced (which adds the Software Design Specification). Each stage of IEC 62304 Clause 5, from development planning through software release, produces specific eSTAR deliverables as natural outputs, so a team that follows the standard systematically never needs to create a separate "FDA version" of its documentation. Teams that structure their documentation this way from project start report cutting submission preparation time by 60-70%.
Mapping the Process to the Artifacts
Summary
- Process-to-deliverable mapping - Clear connection between IEC 62304 development activities and FDA eSTAR requirements eliminates duplicate documentation work
- Documentation level determination - Risk-based Basic vs Enhanced requirements dictate 9 or 10 documents based on software safety classification
- The 10 essential documents - Complete list of FDA-required software documentation with specific IEC 62304 source mapping for efficient preparation
- Efficiency through alignment - Single development process following IEC 62304 naturally produces all FDA submission deliverables
- Practical implementation - Strategic documentation structure decisions made early in development reduce submission preparation time by 60-70%
What you'll learn: How to generate FDA eSTAR software documentation directly from your IEC 62304 development process, eliminating redundant work and ensuring submission completeness.
Introduction
If your medical device has software, you've likely experienced this frustration: your team follows IEC 62304 diligently throughout development, then scrambles to "translate" months of work into FDA submission documents. Requirements get reformatted. Design documentation gets reorganized. Testing records get repackaged. The same information appears in multiple forms across different documents.
This duplicate documentation burden stems from a fundamental misunderstanding about the relationship between IEC 62304 and FDA requirements. IEC 62304 describes a process-based software development lifecycle: what activities to perform and when. FDA's eSTAR software section requires specific deliverable documents: concrete outputs that demonstrate safety and effectiveness.
The disconnect creates real costs: weeks of additional documentation effort, delayed submissions, and increased regulatory review cycles when documentation gaps surface. Quality and regulatory teams waste time creating separate "FDA versions" of documentation that already exists in the design history file.
Most companies miss a key insight: IEC 62304 and FDA eSTAR requirements are complementary, not contradictory. Following IEC 62304 systematically produces all required eSTAR documents as natural byproducts of proper development. The mapping is clear, direct, and eliminates redundant documentation when understood correctly.
This guide provides the complete mapping between IEC 62304 development processes and FDA eSTAR deliverables. You'll learn which specific IEC 62304 activities generate each required document, how to structure your documentation to satisfy both requirements simultaneously, and practical strategies for documentation efficiency that reduce submission preparation time. For how the eSTAR form is organized and what reviewers look for, see our guide chapter on understanding eSTAR requirements.
What's the difference between Basic and Enhanced documentation levels?
FDA recognizes that software risk varies across medical devices. A blood glucose meter carries different patient risk than an implantable pacemaker. Documentation requirements should match this risk profile rather than imposing one-size-fits-all burdens.
Documentation Level Evaluation
The first document FDA requires is your Documentation Level Evaluation: a statement justifying whether your submission requires Basic or Enhanced level documentation. This determination shapes your entire documentation package.
According to the FDA's 2023 Software Documentation Guidance, the level depends on one critical question: Could a failure or flaw of your device software function present a probable risk of death or serious injury to a patient, user, or others in the environment of use?
This assessment must occur prior to implementation of risk control measures. You evaluate the raw, unmitigated risk. If the answer is yes, Enhanced documentation applies. If no, Basic documentation suffices.
Your IEC 62304 software safety classification (Class A, B, or C per Section 4.3) provides the foundation for this determination, but FDA's assessment considers the complete risk picture including reasonably foreseeable misuse and cybersecurity vulnerabilities.
Scope Difference Between Levels
Basic Level requires 9 documents and applies when software failures would not present probable risk of death or serious injury. Examples include non-contact thermometers, blood pressure monitors, and certain diagnostic aids.
Enhanced Level requires 10 documents and applies when software failures could present probable risk of death or serious injury. Examples include ventilators, infusion pumps, pacemakers, and blood screening tests. FDA also recommends Enhanced documentation for all Class III devices and drug/device combination products unless you provide detailed rationale otherwise.
The difference between levels extends beyond document count:
Enhanced Level Requirements Include:
- Software Design Specification (SDS) - Detailed technical design documentation showing how requirements are implemented
- Complete configuration management and maintenance plan documents - Full plans rather than summaries
- Unit and integration level test protocols and reports - Detailed test documentation with expected vs actual results, not just summaries
These additional requirements reflect the higher risk and the corresponding need for FDA reviewers to understand implementation details, not just high-level design and system testing.
Understanding your documentation level determines resource planning, timeline estimates, and documentation strategy from project inception.
The 10 Required eSTAR Software Documents
FDA's eSTAR form organizes software documentation into specific sections. Each serves a distinct purpose in demonstrating safety and effectiveness, with clear origins in your IEC 62304 process.
Document 1: Documentation Level Evaluation
Purpose: Justify Basic vs Enhanced documentation level based on risk analysis
Content Requirements:
- Statement of determined documentation level
- Risk-based rationale accounting for device's intended use
- References to supporting documentation (Risk Management File, Software Description)
IEC 62304 Source:
- Section 4.3 (Software safety classification)
- Section 7.1 (Risk management activities)
- Risk Management File per ISO 14971
Key Elements: Classification rationale, hazard analysis showing software contribution to hazardous situations, patient impact assessment prior to risk controls
Practical Tip: Document this early and thoroughly. FDA may question Basic level determinations for devices with any potential for serious patient harm.
Document 2: Software Description
Purpose: High-level overview enabling FDA reviewers to understand software features, functions, and operational environment
Content Requirements:
- Significant software features and functions
- Operational environment and hardware platforms
- Software inputs and outputs with sources and destinations
- User interactions and clinical workflow
- Block diagrams, flow charts, state diagrams as needed
IEC 62304 Sources:
- Section 5.1 (Software development planning)
- Section 5.3 (Software architectural design)
Key Elements: System context diagrams, deployment architecture, user workflows, interoperability specifications, AI/ML methodology if applicable
Practical Tip: This becomes your submission "executive summary" for software. Invest time making it clear and comprehensive. Reviewers read this first.
Document 3: Risk Management File
Purpose: ISO 14971-compliant analysis demonstrating systematic identification and mitigation of software-related risks
Content Requirements:
- Risk Management Plan with acceptability criteria
- Risk Assessment (analysis, evaluation, control, benefit-risk)
- Risk Management Report showing plan implementation and residual risk acceptability
- Traceability from hazards to risk controls to verification
IEC 62304 Sources:
- Integrated throughout development
- Section 7 (Software Risk Management Process)
- Clause 4.3 (Software safety classification)
Key Elements: Hazardous situations, risk controls with verification, traceability matrix linking hazards → requirements → design → tests
Document 4: Software Requirements Specification (SRS)
Purpose: Complete, testable, traceable requirements defining what the software will do
Content Requirements:
- Functional requirements defining software behaviors
- Performance requirements with specific metrics
- Interface requirements (hardware, software, user)
- Safety-related requirements derived from risk assessment
- Security requirements addressing cybersecurity risks
- All ranges, limits, defaults, and specific values
IEC 62304 Source:
- Section 5.2 (Software requirements analysis)
Key Elements: Requirements with unique IDs, acceptance criteria, traceability to system requirements and risks, verification method for each requirement
Practical Tip: Each requirement must be verifiable through testing. Avoid ambiguous language. Requirements are typically written using "shall" statements with measurable criteria.
Document 5: System and Software Architecture Design Chart
Purpose: Detailed system structure showing component relationships, interfaces, and data flows
Content Requirements:
- Architecture diagrams showing modules, layers, and interfaces
- Module relationships and dependencies
- Data inputs/outputs and flow among modules
- User and external product interactions
- IT infrastructure and deployment architecture
IEC 62304 Source:
- Section 5.3 (Software architectural design)
Key Elements: Component diagrams, sequence diagrams, interface control documents, deployment diagrams, cybersecurity architecture
Note: The diagram in this post shows how "Analysis of software contributing to hazardous situations" bridges architectural design to multiple deliverables. It feeds Software Description, Risk Management File, AND Documentation Level Evaluation. This analysis is foundational for safety classification.
Practical Tip: Consider using standard notation (UML, SysML) for consistency. Ensure diagrams are readable without excessive magnification. Multiple complementary views often work better than one complex diagram.
Document 6: Software Design Specification (SDS) [Enhanced Level Only]
Purpose: Detailed technical implementation showing how design implements requirements
Content Requirements:
- Technical design details of software units/modules
- Algorithms and data structures
- Implementation specifications
- Traceability showing SDS completely and correctly implements SRS
IEC 62304 Source:
- Section 5.4 (Software detailed design)
Key Elements: Unit-level design, detailed algorithms, interface implementations, data structure definitions
Important: Required only for Enhanced (Class C) documentation level. For Basic level, maintain this in your DHF but don't submit unless FDA requests during review.
Practical Tip: SDS should be created prospectively to guide development, not documented retrospectively. FDA looks for evidence that design preceded implementation, not ad hoc coding followed by documentation.
Document 7: Development and Configuration Management
Purpose: Demonstrate controlled development environment with systematic processes
Content Requirements:
For Basic Level:
- Summary of lifecycle processes, coding standards, methods, and tools
- Main deliverables from development activities
- Traceability processes linking requirements → design → tests → risk controls
- Configuration and change management summary
- Software maintenance processes with regression analysis
For Enhanced Level:
- Basic level content PLUS
- Complete configuration management plan document
- Complete maintenance plan document
- Detailed procedures and tools documentation
IEC 62304 Sources:
- Section 5.1 (Software development planning)
- Section 8 (Software configuration management)
- Section 6 (Software maintenance process)
Key Elements: Development plan, configuration management plan, change control procedures, traceability methodology, regression testing approach
Alternative Approach: You may provide Declaration of Conformity to specific clauses of FDA-recognized IEC 62304 instead of narrative documentation. Basic level requires conformity to clauses 5.1.1, 5.1.2, 5.1.3, 5.1.6, 5.1.7, 5.1.8, 5.1.9, Clause 6, and Clause 8. Enhanced requires conformity to complete Clause 5.1, Clause 6, and Clause 8.
Practical Tip: Reference existing QMS procedures where applicable rather than duplicating documentation.
Document 8: Software Testing Documentation
Purpose: Verification and validation evidence demonstrating requirements are met
Content Requirements:
For Basic Level:
- Summary of unit, integration, and system testing
- System level test protocol with expected results, actual results, pass/fail determination
- System level test report demonstrating acceptable execution
- Version tested and overall pass/fail results
- Regression analysis and testing results
- Response to failed tests
For Enhanced Level:
- Basic level content PLUS
- Unit level test protocols and reports (expected vs actual, pass/fail)
- Integration level test protocols and reports (expected vs actual, pass/fail)
IEC 62304 Sources:
- Section 5.5 (Software unit implementation and verification)
- Section 5.6 (Software integration and integration testing)
- Section 5.7 (Software system testing)
Key Elements: Test plans, test cases, test results, requirements traceability matrix, regression test results, anomaly tracking
Critical: Must demonstrate complete requirements coverage. FDA expects traceability from requirements through test cases to results.
Document 9: Software Version/Revision Level History
Purpose: Complete record of tested and released versions
Content Requirements:
- History of all tested versions beginning when design controls applied
- Date, version number, and description of changes for each version
- Final release version with any differences from tested version
- Assessment of differences' effect on safety and effectiveness
- For modifications: highlight prior cleared/approved versions and submission numbers
IEC 62304 Sources:
- Section 5.8 (Software release)
- Section 8 (Software configuration management)
Key Elements: Version numbering scheme, release notes, verification status for each version, change descriptions
Practical Tip: Maintain this from project start, not retroactively. Line-item tabulation format works well. Last entry should be the final release version.
Document 10: Unresolved Software Anomalies
Purpose: Known bugs with safety/effectiveness impact analysis
Content Requirements:
- Description of each unresolved anomaly
- How anomaly was discovered and root cause if known
- Evaluation of impact on safety and effectiveness
- Risk-based rationale for not correcting the anomaly
- Communication to users about mitigations or workarounds
IEC 62304 Sources:
- Section 9 (Software problem resolution process)
Key Elements: Anomaly list with severity classification, impact assessment aligned with risk management plan, rationale for deferring fixes, user communications
Important: Empty list is acceptable if no unresolved anomalies exist. Don't feel compelled to list trivial issues.
Practical Tip: Consider using ANSI/AAMI SW91 defect classification taxonomy for consistency and clarity.
Beyond Software: Cybersecurity Documentation Requirements
The 9-10 software documents covered in this guide address the software development lifecycle and system design requirements. However, FDA's cybersecurity expectations extend beyond software engineering documentation.
Medical device manufacturers must also submit 12-14 cybersecurity-specific documents covering threat modeling, vulnerability management, security architecture, penetration testing results, and security controls implementation. These cybersecurity requirements are separate from (but complementary to) the software lifecycle documentation.
The cybersecurity documentation demonstrates your systematic approach to identifying, analyzing, and mitigating security risks throughout the device lifecycle. This includes premarket security risk assessment, security architecture documentation, and plans for post-market vulnerability management. Our guide covers each of these artifacts document by document, starting with the cybersecurity story FDA expects your submission to tell.
For comprehensive background on cybersecurity requirements:
- Documents FDA Requires for Software and Cybersecurity - Complete list of both software and cybersecurity submission requirements
- Complete Guide to FDA Cybersecurity Risk Assessment - Detailed methodology for security risk analysis
- FDA Cybersecurity Requirements for Medical Devices - Overview of regulatory expectations
- 2025 Update to FDA Cybersecurity Guidance - Latest regulatory developments
While this guide focuses on the IEC 62304-to-eSTAR software documentation mapping, understanding cybersecurity requirements is equally critical for submission success. The software and cybersecurity documentation packages work together to demonstrate comprehensive device safety and effectiveness.
How does IEC 62304 Clause 5 map to eSTAR documents?
IEC 62304 Clause 5 defines the software development process in eight stages (5.1 through 5.8). Understanding how each stage produces eSTAR deliverables reveals the natural alignment between process and documentation.
The Development Process Flow
IEC 62304 structures development as a sequential yet iterative process:
5.1 Software Development Planning → Establishes the development approach, resources, and plans
5.2 Software Requirements Analysis → Defines what the software shall do
5.3 Software Architectural Design → Determines how software will be structured
5.4 Software Detailed Design → Specifies implementation details for software units
5.5 Software Unit Implementation and Verification → Codes and verifies individual units
5.6 Software Integration and Integration Testing → Combines units and tests interfaces
5.7 Software System Testing → Validates complete software against requirements
5.8 Software Release → Prepares software for delivery
Each stage has defined inputs, activities, and outputs. Those outputs become your eSTAR documents.
Direct Process-to-Document Mapping
| IEC 62304 Process Stage | eSTAR Documents Produced |
|---|---|
| 5.1 Development Planning | - Software Life Cycle Process Description - Development & Configuration Management |
| 5.2 Requirements Analysis | - Software Requirements Specification |
| 5.3 Architectural Design | - Software Description - System and Software Architecture Design Chart - Risk Management File (architectural analysis feeds risk assessment) |
| 5.4 Detailed Design | - Software Design Specification (Enhanced only) |
| 5.5 Unit Implementation | - Development & Configuration Management - Software Testing Documentation (unit tests) |
| 5.6 Integration Testing | - Software Testing Documentation (integration tests) |
| 5.7 System Testing | - Software Testing Documentation (system tests) |
| 5.8 Software Release | - Software Version History - Unresolved Software Anomalies |
The Critical Integration Point
This diagram highlights something crucial that many teams miss: "Analysis of software contributing to hazardous situations" serves as a bridge between IEC 62304 Section 5.3 (Architectural Design) and multiple eSTAR deliverables.
This analysis:
- Informs the Software Description by identifying safety-critical functions requiring explanation
- Populates the Risk Management File with software-specific hazards and hazardous situations
- Justifies the Documentation Level Evaluation by demonstrating software contribution to patient risk
This integration point is where software architecture decisions meet patient safety assessment. Getting it right ensures consistency across your submission and demonstrates systematic safety thinking.
Key Insight: No Duplicate Documentation Needed
When you follow IEC 62304 systematically, all required eSTAR documents emerge as natural outputs. You're not creating separate "FDA documentation." Instead, you're properly organizing and presenting the design history file content that regulatory development requires anyway.
The secret is intentional documentation structure from project start, not retroactive reorganization at submission time.
Practical Implementation Strategies
Understanding the mapping intellectually differs from implementing it efficiently. These strategies reduce submission preparation time while maintaining compliance.
Strategy 1: Design Your Documentation Structure Early
Create templates aligned with both IEC 62304 and eSTAR requirements during project planning, not submission preparation.
Implementation:
- Develop SRS template with sections mapping to FDA expectations (functional requirements, performance requirements, safety requirements, security requirements)
- Structure risk management file to support both ISO 14971 and FDA submission needs
- Establish consistent ID schemes enabling traceability (REQ-XXX, DESIGN-XXX, TEST-XXX, RISK-XXX)
- Create traceability matrix template linking requirements → design → tests → risks
Benefit: Reduces submission preparation time by 60-70%. Teams report finishing submissions weeks earlier with pre-structured documentation.
Example: Instead of creating requirements in developer-friendly format then reformatting for FDA, write requirements in FDA-compatible format from day one. It satisfies both needs simultaneously.
Strategy 2: Maintain Living Documents
Update documentation as development progresses rather than batch-documenting at project end.
Implementation:
- Establish documentation review in your development workflow (e.g., requirements reviewed before design, design reviewed before coding)
- Use version control for all documentation, not just code
- Schedule regular documentation reviews (weekly or biweekly) to catch gaps early
- Make documentation updates part of "definition of done" for development tasks
Benefit: Always submission-ready. Eliminates the scramble to reconstruct decisions made months earlier. Supports faster review cycles when FDA questions arise.
Reality Check: Yes, this requires discipline. But retroactive documentation takes longer, introduces errors, and creates DHF issues if documentation doesn't match actual development timing.
Strategy 3: Leverage Your Quality Management System
Many eSTAR requirements map to existing QMS procedures. Reference rather than duplicate.
Implementation:
- For configuration management, reference your QMS configuration control procedure with section numbers
- For change management, reference your design change control SOP
- For development processes, reference relevant QMS procedures (design controls, verification, validation)
- Create summary documents that reference detailed procedures rather than reproducing them
Benefit: Reduces documentation volume while maintaining compliance. Avoids maintaining duplicate procedure sets.
Example: Rather than writing a 20-page configuration management plan, provide a 2-page summary referencing QMS procedures: "Configuration management follows [Company] SOP-123 Design Control, Section 4.5. Version control implemented per SOP-456 Document Control, Section 3.2."
Strategy 4: Create Bidirectional Traceability
FDA reviewers look for complete traceability throughout the documentation package. Implement it systematically.
Implementation:
- Requirements trace forward to design, code, and tests
- Requirements trace backward to system requirements and risks
- Tests trace back to requirements they verify
- Risk controls trace to requirements that implement them and tests that verify them
- Use tools to automate traceability matrix generation where possible
Benefit: Demonstrates completeness. Enables impact analysis for changes. Supports efficient FDA responses when reviewers question specific requirements or test coverage.
Tool Tip: Traceability matrices with thousands of entries become unmanageable in spreadsheets. Consider requirements management tools with built-in traceability if your software complexity warrants it.
Common Pitfalls to Avoid
Learning from others' mistakes prevents costly delays:
Pitfall 1: Starting Documentation at Development End
- Problem: Retroactive documentation doesn't match actual development sequence, creates DHF concerns
- Solution: Establish documentation expectations before coding starts
Pitfall 2: Creating Separate "FDA" and "IEC 62304" Documents
- Problem: Duplicate effort, risk of contradictions, difficult to maintain
- Solution: Single documentation set satisfying both standards through intentional structure
Pitfall 3: Inadequate Traceability Between Documents
- Problem: FDA questions about requirements coverage, risk control verification, test completeness
- Solution: Implement traceability matrices linking all elements from project start
Pitfall 4: Forgetting Documentation Level Rationale
- Problem: FDA questions Basic level determination, requests Enhanced documentation mid-review
- Solution: Document level rationale thoroughly with risk assessment support
Pitfall 5: Missing Safety-Security Integration Points
- Problem: Software architecture doesn't connect to risk assessment, hazardous situation analysis incomplete
- Solution: "Analysis of software contributing to hazardous situations" as explicit architectural design activity feeding risk file
Conclusion
The perceived disconnect between IEC 62304's process focus and FDA's deliverable focus dissolves when you understand the mapping. IEC 62304's development process naturally produces the documents FDA requires for eSTAR submissions.
Key Takeaways:
-
Two Documentation Levels: Enhanced (10 documents) applies when software failures could cause death or serious injury; Basic (9 documents) applies otherwise. Enhanced requires SDS plus more detailed configuration management and testing documentation.
-
Direct Process Mapping: Each IEC 62304 Clause 5 stage produces specific eSTAR documents. Following the standard systematically generates all required deliverables.
-
Single Development Process: You don't need separate "FDA documentation" and "IEC 62304 documentation." Intentional structure satisfies both simultaneously.
-
Critical Integration: "Analysis of software contributing to hazardous situations" bridges architectural design to Software Description, Risk Management File, and Documentation Level Evaluation.
Practical Next Steps:
- Determine Your Documentation Level and document the risk-based rationale thoroughly
- Map Your Existing Documentation to eSTAR requirements to identify gaps
- Establish Templates and Traceability for future projects aligned with both standards
- Implement Living Documentation practices to stay submission-ready continuously
Medical device software documentation doesn't have to be painful. When IEC 62304 and FDA requirements align through proper planning, documentation becomes a natural byproduct of good development practice rather than a burdensome afterthought. The key is understanding the mapping and implementing it systematically from project inception.
References
-
FDA, "Content of Premarket Submissions for Device Software Functions," FDA Guidance, June 2023. [Online]. Available: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/content-premarket-submissions-device-software-functions
-
IEC 62304:2006+A1:2015, "Medical device software — Software life cycle processes" [Online]. Available: https://www.iso.org/standard/38421.html
-
ISO 14971:2019, "Medical devices — Application of risk management to medical devices" [Online]. Available: https://www.iso.org/standard/72704.html
-
FDA, "General Principles of Software Validation," FDA Guidance, January 2002. [Online]. Available: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/general-principles-software-validation
This content is for educational purposes and represents interpretation of current regulatory guidance. Always consult with qualified regulatory professionals for specific compliance decisions.
Frequently asked questions
›How many software documents does FDA require in an eSTAR submission?
Nine documents for Basic documentation level and ten for Enhanced. Enhanced adds the Software Design Specification plus complete configuration management and maintenance plans and unit and integration test protocols with reports, rather than summaries.
›How do I know if my device needs Basic or Enhanced documentation?
It hinges on one question from FDA's 2023 software guidance: could a failure or flaw of the device software function present a probable risk of death or serious injury? You evaluate the raw, unmitigated risk before risk controls. Yes means Enhanced, no means Basic. FDA also recommends Enhanced for all Class III devices and drug/device combination products.
›Do I need separate documentation for IEC 62304 and FDA?
No. Following IEC 62304 systematically produces all required eSTAR documents as natural outputs of development. The trick is intentional documentation structure from project start, so a single documentation set satisfies both.
›Which IEC 62304 clauses map to which eSTAR documents?
Clause 5.2 (requirements analysis) produces the SRS, 5.3 (architectural design) produces the Software Description and architecture chart, 5.4 (detailed design) produces the SDS for Enhanced level, 5.5 through 5.7 produce the testing documentation, and 5.8 (release) produces the version history and unresolved anomalies list.
›Does the eSTAR software documentation cover FDA's cybersecurity requirements too?
No. The 9-10 software documents cover the software lifecycle. Manufacturers must also submit 12-14 cybersecurity-specific documents covering threat modeling, security architecture, penetration testing results, and security controls. The two packages are separate but complementary.